
File
Open - Read molecule from file: after the "Open File" dialog opens make sure you select the proper format before selecting the file to open. If you use the "All files - *.*" filter the program will try to guess the file format, which is not usually what you want except for pdb, xyz and other well known file formats. For a list of supported file formats have a look at the file formats section.
Save - Save selected molecule to file.
Save Image - Take snapshot of 3D view and save to TIFF or PNG file.
Save to PostScript - Save content of 3D view to PostScript format; transparent geometry will not be exported correctly.
- When saving to PostScript a
psextension is automatically added to the file. - For best results when exporting molecules with atoms rendered as spheres select Display->Molecule->Atom->gluSphere.
Save to PDF - Save content of 3D view to PDF format; transparent geometry will not be exported correctly.
- When saving to PDF a
pdfextension is automatically added to the file. - For best results when exporting molecules with atoms rendered as spheres select Display->Molecule->Atom->gluSphere.
Edit
Unselect - Unselect all selected objects (atoms, bonds, residues, molecules).
Delete Molecule - Delete selected molecule. Asks for confirmation.
Clear - Delete all molecules. Asks for confirmation.
Reset Molecule - Resets selected molecule's position and orientation. Enabled only when a molecule is selected.
Reset Camera - Resets viewpoint.
Display
Molecule - Open the Molecule Display Settings dialog. Use this dialog to change the atom, bond and residue rendering style and to hide/show the dipole moment (if present).
Background - Change background color.
3D View Properties
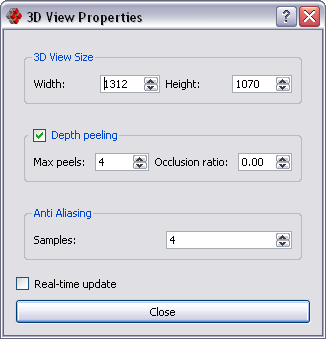
Change 3D View properties.
- 3D View Size: changes the 3d view size.
- Depth peeling: these parameters affect the rendering of transparent surfaces:
- when rendering multiple transparent surfaces increase the Max peels parameter until you can see all the overlapping transparent surfaces
- change the Occlusion ratio parameter to increase rendering speed or eliminate rendering artifacts:
0= best quality,1= max speed
- Anti Aliasing: increase the number of samples for better looking pictures with little or no jaggies(i.e. steps in diagonal lines or edges); note that each sample is a complete redraw, so rendering time will increase linearly with the number of samples
Show/Hide Plane Probe - Show/Hide plane probe: when the plane is visible the menu item is set to "Hide", to "Show" otherwise. The plane probe can be used to visualize a scalar field (e.g. Electrostatic Potential) and to display the value of the field at a specific point in space by moving the crosshair cursor on the plane. Plane probe interaction:
- Move plane: middle mouse button (left on Mac OS)
- Move cursor on plane: left mouse button (currently disabled on Mac OS)
- Change window and center point used for color mapping: right mouse button
- Change window: move mouse along Y axis
- Change center point: move mouse along X axis
Show/Hide MEP Scalar Bar - Show/Hide color bar associated with Electrostatic Potential; use this feature only after a Molecular Electrostatic Potential has been computed and mapped on a surface.
Show/Hide Probe Scalar Bar - Show/Hide color bar associated with Plane Probe; use this feature only after the data to visualize have been generated through the Analysis-Plane Probe dialog.
Show/Hide Axes - Show/Hide Axes at the bottom right corner of the 3D View.
Show/Hide Bounding Boxes - Show/Hide selection boxes.
Shaders - Open Shaders dialog; enabled only if a molecule is selected and the system supports OpenGL 2.0 shaders.
Interaction
Used to dispatch mouse events to camera or selected molecule.
Camera - Move/Rotate camera.
Molecule - Move/Rotate selected molecule. Only enabled when a molecule is selected.
Pick Atom/Bond - Enable/Disable picking of individual atoms/bonds/residues. Only enabled when a molecule is selected and Iteraction-Molecule is checked. Information about the selected object is displayed in the status bar.
Event Recorder - Opens the event recording window which allows to save GUI events to a file - Used for testing purpose only.
Event Player - Opens the event player window which allows playback GUI events read from file - Used for testing purpose only.
Animation
Start/Stop Animation - Start/Stop animation: starts/stops the animation timer; animation advances to next frame each time the animation timer fires.
Next - Advance to next frame. Disable if animation timer started.
Previous - Move to previous frame. Disabled if animaiton timer started.
Timestep - Change animation time step.
Per-Molecule Settings - Open Molecule Animation Settings dialog to change animation settings for the selected molecule, see animation settings section for details.
Export Animation - Export animation to individual frames or AVI (Windows only). By default all the visible objects are animated and exported, including any visible surface.
Surfaces
Electron Density - Generate surface(s) from electron density data: molecular orbitals or density matrix.
Grid Data - Generate surface from data read from Gaussian cube files.
Solvent Accessible Surface - Generate SAS as iso-surface.
Solvent Excluded Surface - Generate SES (Connolly) surface using an internal algorithm (points only) or M.F. Sanner MSMS program (highly recommended, very fast); you can get it here.
Surfaces menu items are enabled only when a molecule is selected.
It is possible to color-code the generated surfaces with molecular electrostatic potential values if the information is available.
Analysis
Analysis menu items are enabled only when a molecule is selected.
Plane Probe - Open the Plane Probe dialog used to generate the data that will be visualized on the surface of the plane probe.
Distance - Compute distance between two atoms, enabled when two atoms are selected.
- unselect all (Ctrl/Command - U)
- select two atoms while holding the Shift key down to enable multiple selection
- click on Analysis-Distance: the distance between the two selected atom will be displayed in the status bar
Angle - Compute angle among three atoms, enabled when three atoms are selected.
- unselect all (Ctrl/Command - U)
- select three atoms while holding the Shift key down to enable multiple selection
- click on Analysis-Angle: the angle among the three selected atom will be displayed in the status bar
Dihedral Angle - Compute dihedral angle, enabled when four atoms are selected.
- unselect all (Ctrl/Command - U)
- select four atoms while holding the Shift key down to enable multiple selection
- click on Analysis-Dihedral Angle: the dihedral angle be displayed in the status bar
Note 1: Each time an atom is added to the selection list the Added to selection message appears in the status bar
Note 2: Following the instructions above works fine but the first atom added to the selection list is not highlighted; to highlight the first atom too select the molecule first and then start adding atoms into the selection list
Radiation Spectrum - Display infra-red or Raman activity spectrum.
Help
Documentation - Open on-line copy of Molekel documentation. Opens local copy in Molekel < 5.1.
Molekel Web Page - Open Molekel website url in external browser.
License - Display license and copyright information.
About - Display version and copyright information.